AlgPred:
PREDICTION OF ALLERGENIC PROTEINS AND MAPPING OF IgE EPITOPES
Sudipto Saha and G. P. S.
Raghava*
Institute of Microbial
Technology, Sector-39A, Chandigarh, India
Running
Tite: Allergens prediction
Address correspondence to:
Dr. G. P. S. Raghava, Scientist, Bioinformatics Centre Institute of Microbial
Technology Sector 39A,
Chandigarh, INDIA, Phone: +91-11-26907444; Fax: +91-172-2690632 E-mail: raghava@iiitd.ac.in
Available at :
http://webs.iiitd.edu.in/raghava/algpred
Algorithm used in developing AlgPred server
Data set partitioning
One of the challenges in developing any prediction method is to minimize the similarity between proteins used for training and protein used for testing. Though removing redundancy is not a problem but it reduces the number of proteins used for training, which is not good for any learning method. In this study we a different approach have been used to minimize similarity between proteins used for training and testing without reducing total number of proteins. First proteins are clustered based on similarity using BLAST E-value 8E-4 (26% identity for one sequence pair). These clusters were divided into five sets in such a way that each set have nearly equal number of sequences, where all proteins of a given cluster are kept in one set. As sequences in one cluster do not have similarity (e-value of 8E-4) with sequences of other clusters so sequences in one set will not have similarity with sequences of other sets.
Presence
of IgE epitopes
In this
approach, a protein is predicted allergen if protein have one or more than one
IgE epitopes. Thus in this study,
similarity based approach has been used, where a protein is assigned allergen
if it have a region/peptide identical to known IgE epitopes. First all 183 IgE epitopes scanned against
all proteins in dataset and found 61 hits in allergens and 16 hits in
non-allergens, when stringent condition of 100% identity was used. Ideally, IgE
epitope should not be present in non-allergens, thus epitopes present in
non-allergens has been analyzed. It has been observed that most of the epitopes
founds in non-allergen were short in length (3 or 4 residues); for example
epitopes HWR, IRRA and YHVP have 7, 6 and 1 hits respectively in non-allergens.
Due to their short length they were unable to provide specificity. Thus for
further study, only those epitopes (178 epitopes) have been used which have
five or more than five residues. As shown in Table 1, for PID 100 we got 56
hits in allergen (or 9.69 % of allergens were correctly assigned as allergens)
and 2 hits in non-allergens (or 0.28% of non-allergens assigned wrongly as
allergens). In order to increase the sensitivity or percent coverage of
allergens we relaxed the criteria. Though the sensitivity increased but the
percent of false assignment of non-allergen to allergen also increased when we
relaxed the criteria. Thus, we set PID
cut-off based on length of epitope rather than uniform cut-off. This way we
were able to achieve better sensitivity without loosing significant
specificity. The best results were obtained using PID865 where PID cut-off is
80, 60 and 50 for epitopes having residues less than 10, between 10 to 15 and
more than 15, respectively.
Table 1: Searching of 178 IgE epitopes in protein dataset consists
of 578 allergens and 700 non-allergens. Different percent identity (PID)
cut-off was used to search epitopes in proteins, cut-off was also set based on
amino acids (A.A.) in IgE epitopes.
|
Approach |
PID (Cut-off) |
Total Hits |
|
|
Allergens |
Non-allergens |
||
|
PID100 |
100 |
56 (9.69%) |
2 (0.28%) |
|
PID81 |
>80 |
77 (13.32%) |
8 (1.11%) |
|
PID80 |
³80 |
102 (17.65%) |
74 (10.57 %) |
|
PID876 |
>80 (epitopes have
£ 9 A.A.) ³70 (epitopes have > 9 & £ 15 A.A.) >60 (epitopes
have > 15 A.A.) |
91 (15.74%) |
11 (1.57%) |
|
PID865 |
>80 (epitopes have
£ 9 A.A.) ³60 (epitopes have > 9 & £ 15 A.A.) >50 (epitopes
have > 15 A.A.) |
101 (17.47%) |
13 (1.85%) |
Allergen Representative Peptides collection
and Prediction using ARPs
The
dataset of ARPs consists of 2890 ARPs (24 amino acid peptides) obtained from
Bjorklund et al., 2005 . They collected high-quality repositories of amino acid
sequences of proteinaceous allergens (allergen database) and non-allergens
(consumed commodities, such as rice, apple, tomato etc.) and generated all
possible overlapping 24mer peptides for both types of proteins (allergen and
non-allergens). Based on the global similarity scores of each allergen peptide,
a set containing 2890 ARPs were created which has high similarity in allergenic
proteins but not in non-allergenic proteins.
The
approach was tested on 664 allergens and 700 non-allergens . As shown in Table
2, the number of correctly assigned allergens (or sensitivity) increased from
52.71% to 94.28% when e-value BLAST increased from 10-9 to 1.0. Though the sensitivity
increased with increase in e-value, the number of non-allergen falsely
predicted allergens also increased from 2 to 338. It is found that e-value of 0.001 provides reasonably high
sensitivity 83.58% with only 15 false positive (non-allergens predicted as
allergens). Thus e-value 0.001 used as default cut-off for further study.
Table 2 : The search results of 1364 proteins
(664 allergens & 700 non-allergens), which
searched against ARPs database using BLAST.
|
E-value |
Total Hits
|
|
Allergen
|
Non-allergen
|
|
|
1 |
626
(94.28%) |
338
(48.25%) |
|
10-1 |
586
(88.22%) |
48
(6.86%) |
|
10-2 |
562
(84.64%) |
23
(3.29%) |
|
10-3 |
555 (83.58%) |
15 (2.14%) |
|
10-4 |
527
(79.37%) |
8 (1.14%) |
|
10-6 |
465
(70.03%) |
5 (0.71%) |
|
10-9 |
350
(52.71%) |
2
(0.28%) |
MEME/MAST
MEME/MAST: version 3.0.4, obtained from http://meme.sdsc.edu/meme/ website. MEME
(Multiple Em for Motif Elicitation) is a tool for discovering motifs in a group
of related protein sequences. A motif is a sequence pattern that occurs
repeatedly in a group of related protein sequences. MEME represents motifs as
position-dependent letter-probability matrices, which describe the probability
of each possible letter at each position in the pattern. MEME takes as input a
group of protein sequences (the training set) and output as many motif as
requested. MEME uses statistical modeling techniques to automatically choose
the best width, number of occurrences, and description for each motif. MAST
(Motif Alignment and Search Tool) is a tool for searching biological sequence
databases for sequences that contain one or more of a group of known motifs.
MAST takes as input a file containing the descriptions of one or more motifs
and searches sequence databases that have been created that match the motifs.
First, five MEME matrices have been created corresponding to five sets, one
matrix for one set. Then each matrix
was used in as input file for searching motifs in remaining four sets using
program MAST. Finally we compute the performance of this approach and achieved
the sensitivity in the range of 7% (at 0.001 e-value) to 94% (at 100 e-value)
(Table 3). Though the sensitivity increased with increase in e-value but the
percent of wrong assignment of non-allergens to allergens also increased from
2.85% to 66.86%. This demonstrate that motif based approach developed in this
study have low specificity.
Table 3:
MEME/MAST results of allergen and
non-allergen motifs. It shows allergen hits out of total 578 allergens and
non-allergen hits out of total 700 non-allergens.
|
E-value |
Total Hits
|
|
Allergen
|
Non-allergen
|
|
|
10-3 |
38
(6.57%) |
20
(2.86%) |
|
10-1 |
86
(14.88%) |
62
(8.86%) |
|
1 |
142
(24.57%) |
113
(16.14%) |
|
10 |
246
42.56%) |
240
(34.29%) |
|
20 |
309
(53.46%) |
288
(41.14%) |
|
50 |
427
(73.88%) |
389
(55.57%) |
|
100 |
543
(93.94%) |
468
(66.86%) |
Support vector machine
The support vector machines (SVM) are
universal approximator based on statistical and optimising theory. The SVM is
particularly attractive to biological analysis due to its ability to handle
noise, large dataset and large input spaces. The SVM has been shown to perform
better in protein secondary structure, MHC and TAP binder prediction and
analysis of microarray data. The basic idea of SVM can be described as follows;
first the inputs are formulated as feature vectors. Secondly these feature
vectors are mapped into a feature space by using the kernel function. Thirdly,
a division is computed in the feature space to optimally separate to classes of
training vectors. The SVM always seeks global hyperplane to separate the both
classes of examples in training set and avoid overfitting. The hyperplane found
by SVM is one that maximise the separating margins between both binary classes
This property of SVM is made is more superiors in comparison to other
classifiers based on artificial intelligence.
In this study, we have used SVM_light
to predict the
allergenic proteins. The software is freely downloadable from http://www.cs.cornell.edu/People/tj/svm_light/
. The software enable the users to define a number of parameters and allow to
select a choice of inbuilt kernel function including linear, RBF, Polynomial
(given degree) or user defined kernel.
Preliminary tests showed that the radial basis function (RBF) kernel gives results better than other kernels. Therefore, in this work we used the RBF kernel for all the experiments. The input vectors used were amino acid composition (20 vectors) and dipeptide composition (400 vectors) of each protein sequence.
Protein features
Amino acid
composition. Amino acid composition is the fraction of each amino
acid in a protein. The fraction of all 20 natural amino acids was calculated
using the following equations:
Fraction of amino acid i =

where i can be
any amino acid.
Dipeptide
composition. Dipeptide composition was
used to encapsulate the global information about each protein sequence, which
gives a fixed pattern length of 400 (20 ´ 20). This representation encompassed the information about
amino acid composition along local order of amino acid. The fraction of each
dipeptide was calculated using following equation:
fraction of dipep (i) =
![]()
where dipep(i) is one out of 400
dipeptides.
Five fold
cross-validation:
The performance of all methods
developed in this study is evaluated using five-fold cross validation. In
five-fold cross validation dataset has been divided into five sets, where each
set have nearly equal number of allergens and non-allergens. The training and
testing of every method has been carried out five times, each time using one
distinct set for testing and remaining four sets for training. The overall
performance of a method is average performance over five sets.
Performance
measures
The standard parameters have been used to evaluate the
performance of various methods developed in this study. Following is brief
description of the parameters; i) Sensitivity is the percent of epitopes that
are correctly predicted as epitopes also referred as recall; ii) Specificity is
the percent of correctly predicted as non-epitopes; iii) Accuracy is the
proportion of correctly predicted peptides; iv) PPV (positive prediction value)
is the probability of correctly positive prediction also referred as precision v) NPV(negative prediction value) is the
probability of correctly negative prediction and vi) Matthew’s correlation
coefficient (MCC). The parameters may be calculated by the following equations.
![]()
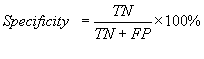
![]()
![]()

MCC=![]()
Where TP and FN refer to true
positive and false negatives, TN and FP refer to true negatives and false
positives.
The
performance of SVM module based on amino acid and dipeptide composition
The following Table 4 and Table 5 demonstrate the performance of SVM based method using amino acid and dipeptide composition respectively. The RBF kernel was used and SVM module based on amino acid composition parameters used are g=50; c=1; and j=1;
Whereas SVM module based on dipeptide composition parameters used are g=100; c=1; and j=1.
Table 4: Performance
of SVM based method using amino acid composition
|
Threshold |
Sensitivity |
Specificity |
Accuracy |
PPV |
NPV |
MCC |
|
1.0 |
0.3374 |
0.9829 |
0.6918 |
0.9417 |
0.6442 |
0.4336 |
|
0.8 |
0.4243 |
0.9700 |
0.7239 |
0.9208 |
0.6729 |
0.4843 |
|
0.6 |
0.5200 |
0.9586 |
0.7608 |
0.9116 |
0.7093 |
0.5456 |
|
0.4 |
0.5878 |
0.9386 |
0.7804 |
0.8871 |
0.7357 |
0.5732 |
|
0.2 |
0.6539 |
0.9243 |
0.8024 |
0.8765 |
0.7657 |
0.6099 |
|
0.0 |
0.7357 |
0.8929 |
0.8220 |
0.8494 |
0.8054 |
0.6422 |
|
-0.2 |
0.8383 |
0.8543 |
0.8471 |
0.8253 |
0.8667 |
0.6930 |
|
-0.4 |
0.8887 |
0.8186 |
0.8502 |
0.8009 |
0.9009 |
0.7053 |
|
-0.6 |
0.9061 |
0.7614 |
0.8267 |
0.7573 |
0.9096 |
0.6680 |
|
-0.8 |
0.9391 |
0.6957 |
0.8055 |
0.7171 |
0.9347 |
0.6441 |
|
-1.0 |
0.9583 |
0.6100 |
0.7671 |
0.6687 |
0.9489 |
0.5933 |
Table 5: Performance of
SVM based method using dipeptide composition
|
Threshold |
Sensitivity |
Specificity |
Accuracy |
PPV |
NPV |
MCC |
|
1.0 |
0.0957 |
1.0000 |
0.5922 |
1.0000 |
0.5742 |
0.2346 |
|
0.8 |
0.1791 |
1.0000 |
0.6298 |
1.0000 |
0.5978 |
0.3275 |
|
0.6 |
0.2696 |
0.9886 |
0.6643 |
0.9509 |
0.6229 |
0.3852 |
|
0.4 |
0.3913 |
0.9686 |
0.7082 |
0.9109 |
0.6602 |
0.4538 |
|
0.2 |
0.5461 |
0.9429 |
0.7639 |
0.8870 |
0.7174 |
0.5441 |
|
0.0 |
0.7043 |
0.9100 |
0.8173 |
0.8654 |
0.7903 |
0.6353 |
|
-0.2 |
0.8278 |
0.8500 |
0.8400 |
0.8193 |
0.8586 |
0.6786 |
|
-0.4 |
0.8922 |
0.7743 |
0.8275 |
0.7645 |
0.8988 |
0.6657 |
|
-0.6 |
0.9374 |
0.6771 |
0.7945 |
0.7046 |
0.9312 |
0.6259 |
|
-0.8 |
0.9600 |
0.5657 |
0.7435 |
0.6449 |
0.9474 |
0.5589 |
|
-1.0 |
0.9757 |
0.4471 |
0.6855 |
0.5918 |
0.9601 |
0.4840 |
Hybrid
Approach
The objective of this approach is to improve the
sensitivity as well as the specificity of allergen prediction method. Each
approach has its own limitations, as some provides high sensitivity but low
specificity and vice verse. In order to get high sensitivity without
loosing much specificity or high specificity with reasonable percent coverage,
we combined two or more than two approaches. First, SVM and IgE epitope based
approaches have been combined, where a protein is assigned as allergen if
predicted allergen by IgE method (PID865) and also have SVM score ³ -0.5. A protein is assigned allegen or non allergen using
SVM approach, if protein have no similarity with known IgE epitopes. As shown
in Table 6, the sensitivity increased around 11% (33.74 to 44.52), where as the
specificity decreased marginally by 0.15%. Similar trend observed when SVM
based method using dipeptide composition has been combined with IgE epitope
based method. No improvement has been observed when motif based and SVM based
approaches have been combined.
Table 6: The performance of
hybrid approach, which combines SVM based approach using amino acid composition
and IgE epitope based approach (PID865).
|
Threshold |
Sensitivity |
Specificity |
Accuracy |
PPV |
NPV |
MCC |
|
1.0 |
0.4452 |
0.9814 |
0.7396 |
0.9517 |
0.6836 |
0.5211 |
|
0.8 |
0.4922 |
0.9700 |
0.7545 |
0.9309 |
0.7000 |
0.5405 |
|
0.6 |
0.5652 |
0.9586 |
0.7812 |
0.9181 |
0.7293 |
0.5829 |
|
0.4 |
0.6191 |
0.9386 |
0.7945 |
0.8922 |
0.7509 |
0.5995 |
|
0.2 |
0.6713 |
0.9243 |
0.8102 |
0.8793 |
0.7749 |
0.6248 |
|
0.0 |
0.7443 |
0.8929 |
0.8259 |
0.8509 |
0.8106 |
0.6499 |
|
-0.2 |
0.8417 |
0.8543 |
0.8486 |
0.8259 |
0.8692 |
0.6963 |
|
-0.4 |
0.8887 |
0.8186 |
0.8502 |
0.8009 |
0.9009 |
0.7053 |
|
-0.6 |
0.9061 |
0.7614 |
0.8267 |
0.7573 |
0.9096 |
0.6680 |
|
-0.8 |
0.9391 |
0.6957 |
0.8055 |
0.7171 |
0.9347 |
0.6441 |
|
-1.0 |
0.9583 |
0.6100 |
0.7671 |
0.6687 |
0.9489 |
0.5933 |
Evaluation on Independent Dataset and on
Swiss-Prot protein sequences
It has
been shown in number of studies that there is biasness in performance of the
method if it is trained and tested on same dataset despite n-fold cross-validation
(37,38). Thus it’s advisable to test any newly developed method on an
independent dataset not used in training or testing of the method. In order to
avoid any biasness we used default parameters for each approach (cut-off etc.).
As shown in Table 7, the accuracy of prediction based on SVM based approaches
were around 85%, followed by ARPs BLAST of 67%. The performance on Swiss-Prot
proteins shows the SVM based method using amino acid and dipeptide composition,
falsely predicted 46.74% and 39.30% non-allergens as allergens respectively.
Though specificity of these SVM based method is poor but same time coverage or
sensitivity is higher than other method. In reverse IgE epitope and MEME
methods predicts low rate of false positive but have poor sensitivity.
Table 7: Performance of different methods on 101725
non-allergens obtained from Swiss-Prot and on 323 allergens (independent
dataset not used in training or testing of methods).
|
Prediction Methods |
101725
Non-allergens obtained from Swiss-Prot |
Independent
dataset of 323 allergens |
|
|
Falsely predicted allergens |
Specificity
(Predicted non-allergens) |
Allergens
correctly predicted allergens (Sensitivity) |
|
|
SVMc |
44684 |
56.07% |
272 (84.21%) |
|
SVMd |
39590 |
61.09% |
274 (84.83%) |
|
MAST (ev100) |
13545 |
86.68% |
58 (17.95%) |
|
MAST (ev 0.1) |
3480 |
96.58% |
40 (12.38%) |
|
BLAST (ARP) |
2060 |
97.97% |
215 (66.56%) |
|
IgE Epitope |
1777 |
98.25% |
35 (10.84%) |
Available at : http://webs.iiitd.edu.in/raghava/algpred