The dataset used in this study to train the SVM was collected from MHCBN and AntiJen.The dataset contained the peptides whose affinity value (IC50) are already determined experimentally.The size of the dataset was reduced by selecting and keeping only peptides, having 9 amino acids length (as nonamers are ideal binders of MHC-I molecules).The redundancy of the dataset was further reduced so that no two peptides have >90% sequence identity.This dataset consists of 402 binders and 222 nonbinders.
Support Vector Machine
The support vector machine (SVM) are universal approximator based on stastical and optimising theory.The SVM is particularly attractive to biological analysis due to its ability to handle noise, large dataset and large input
spaces. The SVM has been shown to perform better in protein secondary structure prediction, MHC and TAP binder prediction and analysis of microarray data. The basic idea of SVM can be described as follows : First the inputs are
formulated as feature vectors. Secondly these feature vectors are mapped into a feature space by using the kernel function. Thirdly a division is computed in the feature space to optimally seperate to classes of training vectors.
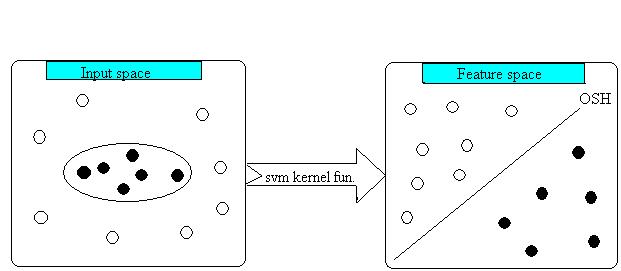
The SVM always seeks global hyperplanes to seperate both classes of example in training set and avoid overfitting. The hyperplane found by SVM is one that maximise the seperating margins between both binary classes. This property
of SVM makes it more superior than other classifiers based on artificial intelligence. The basic idea of SVM is depicted below.
In this study we have used SVM_light to predict the affinity of HLA-A2 peptide binders. The software is freely downlodable from http://www.cs.cornell.edu/People/tj/svm_light/. The software enable the users to
define a number of parameters and allow to select a choice of inbuilt kernel function including linear, RBF, Polynomial (given degree) or user defined kernel.
Evaluation of Modules:- The performance of modules developed in this method is evaluated using leave-one-out approach. In leave-one-out approach one peptide is taken as testset and the rest of them are grouped as trainingset.The training and testing of the module is is carried out
the number of times equal to the number of peptides in the datset, each time using one distinct peptide for testing and the remaining grouped for training.The formula for accuracy calculation is shown below.
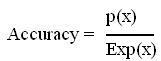
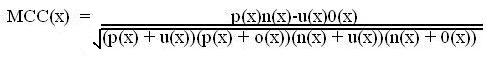
Prediction approaches:- In this study the authors have developed modules on the basis of SVM.The SVM was trained with various inputs to encapsulate the global information of peptide binders and non-binders for more accurate predictions.Modules developed in this study are:
1.Binary based SVM:- The SVM was provided with 20 dimensional vector on the basis of binary pattern of amino acids in the peptides.The SVM was trained for binary patterns of the peptides.The best results were obtained at Linear kernel C = 1.
2.Physico-chemical properties based SVM:- The SVM was developed on the basis of average physico-chemical properties of peptide sequence.The properties of the peptides waere dtermined by considering 36 physico-chemical properties.The best results were obtained at Linear kernel C = 10.
|