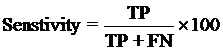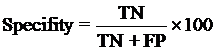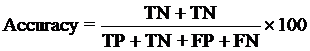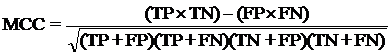| |
MetaPred:A webserver for the Prediction of Cytochrome P450 Isoform responsible for Metabolizing a Drug Molecule |
|
|
Toxipred |KiDoQ | GDoQ | NPTOPE |KetoDrug |CRDD |OSDD |IMTECH | Raghava |
|
|
|
|
Main DataSet :All substrates that are metabolize by any of the following isoform CYP 3A4, 2D6, 1A2, 2C9 and 2C19 were obtained from DrugBank2.5 [11,12]. We got total 372 drug molecules where each molecule metabolized by at least one of the five isoforms. In order to create exclusive dataset, we remove all those molecules that are metabolized by more than one isoforms. Finally, we got a dataset of 216 drug molecules, which consists of 111, 47, 29, 20 and 19 molecules metabolized through CYP 3A4, 2D6, 1A2, 2C9 and 2C19 isoforms respectively.Independent DataSet :We created an independent dataset in order to evaluate performance without any bias. Thus we downloaded 146 molecules from DrugBank where each molecule is reported to be metabolizing by one or more isoform used in this study. This independent dataset consists of total 146 molecules, where 92, 74, 41, 47 and 49 molecules have metabolic specificity for CYP 3A4, 2D6, 1A2, 2C9 and 2C19 isoform respectively. Name of the molecule used in main dataset and independent dataset are given in Supplementary datasheet (Table: S1 - S6).Descriptors calculation :For 2D and 3D QSAR modeling we have used Vlife and Chemistry Development Kit (CDK) software for the calculation of 2D and 3D descriptors.By Vlife and CDK we have received 1002 & 174 descriptors respectively. These descriptors differentiated into different catagory according to there properties like : constitutional descriptors, topological descriptors, connectivity indices, information indices, 2D autocorrelation, burden eigenvalues, topological charge indices,functional groups, molecular properties and eigenvalues based indices.Feature Selection :One of the major challenges in a QSAR is selection of relevant molecular descriptors from large number of descriptors.we have used Weka based GreedyStepWise and genetic search approach for the selection of features (descriptors), to investigate which features are the most predictive ones. Cross-validation Techniques Support Vector Machines:The SVM was implemented using freely downloadable software package SVM_light written by Joachims (Joachims 1999). The software enables the user to define a number of parameters as well as to select from a choice of inbuilt kernal functions, including a radial basis function (RBF) and a polynomial kernal.
Evaluation module:The performance modules constructed in this study were evaluated using a 5-fold cross-validation technique. In the 5-fold cross-validation, the relevant dataset was partoned randomly into five equally sized sets. The training and testing was carried out five times, each time using one distinct set for testing and the remaining four sets for training.The performance of the methods was computed using the following formulas :
Where TP is correctly predicted CYPs substrate, TN is correctly predicted negative substrate as non-substrate for those isoform; FP is the number on non-substrate molecules predicted as substrate and FN is number of substrate molecules predicted as non-substrate. Matthew’s correlation coefficient (MCC) equal to 1 is regarded as a perfect prediction, whereas 0 is for completely random prediction.
|