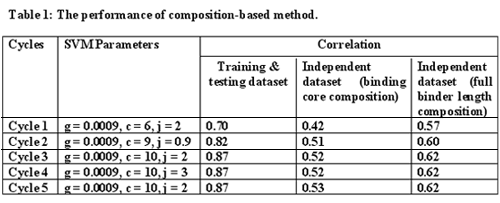
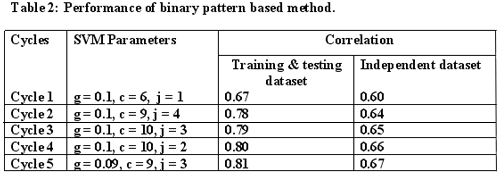
Prediction of binders and binding affinity of peptides binding to MHC class-II molecules is not easy due to the variable length of these binders, but it is known that the binding core is of 9 amino acids only.So, in the present study we have used a new strategy called Support Vector Machine Optimization Technique (SVMOT) to predict the binding core of peptides binding to HLA-DRB1*0401 allele. After finding the binding core we also developed a method to predict the binding affinity of these peptides for this allele.
Dataset:The dataset used in this study was collected fetched from the databases AntiJen and MHCBN and also from the literature. The training dataset contained 659 peptides along with the natural log values of their IC50 (Inhibitory Concetration 50) values. The independent dataset had 50 peptides with the natural log of their IC50 values written besides them.
Amino acid composition:
Amino acid composition is the fraction of each amino acid in a protein. The fraction of all 20 natural amino acids was calculated using the following equation:
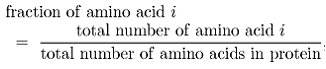
Where 'i' can be any amino acid.
Binary pattern:
Each amino acid is represented by a vector of dimension 20 as unique pattern containing binary numbers(ninteen 0's and a single 1). For example Alanine is represented by 10000000000000000000 and Cysteine by 01000000000000000000.
Support Vector Machine Optimization Technique (SVMOT)
Support Vector Machine Optimization Technique:
Following diagram explains the strategy used by us in developing the method:
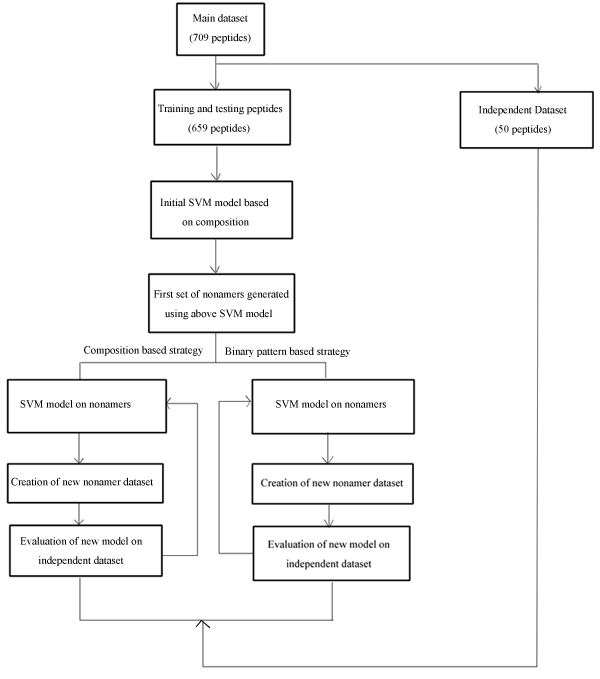 Evaluation of the performance:
Evaluation of the performance: In this study five fold cross validation was used to evaluate the performance of the method. The original dataset was divided randomly into five sets. All of the sets consisted of nearly equal number of peptides. Four sets were used for training and the remaining one for the testing of the methods and this procedure was repeated five times so that each set is used at least once for training as well as for testing.The correlation between the actual and predicted affinity was then calculated by the formula:
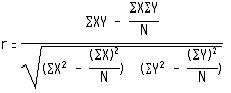 Performance on the independent/blind dataset:
Performance on the independent/blind dataset:
The models developed above were tested on the independent/blind dataset. A blind dataset consists of peptides, which have never been used, in the training as well as in the testing of the methods. The independent dataset used in this method had 50 peptide of varying lengths along with natural log of their experimentally determined IC50 values.