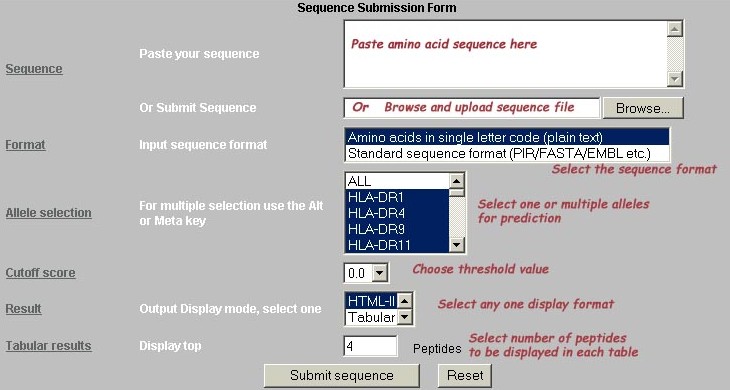
MHC2Pred Help
The MHC2Pred is an SVM based method for prediction of promiscuous MHC class II binding peptides.The average accuracy of SVM based method for 42 alleles is ~80%. The performence of the method was poorer for few allele due to smaller size of dataset. The performence of the method was tested through 5-fold cross-validation. The data for training has been extracted from MHCBN and JenPep database.This method will be useful in cellular immunology, Vaccine design, immunodiagnostics, immunotherapeuatics and molecular understanding of autoimmune susceptibility. The stepwise help for using the server and snapshot of submission page with approriate labelling has been described below:-
 Prediction Results-: The results of the prediction displayed in user-freindly text formats.Each of result display format firstly provides a compehensive account of length of input sequence,prediction approach,nonamers generated and threshold as shown below.
Prediction Results-: The results of the prediction displayed in user-freindly text formats.Each of result display format firstly provides a compehensive account of length of input sequence,prediction approach,nonamers generated and threshold as shown below.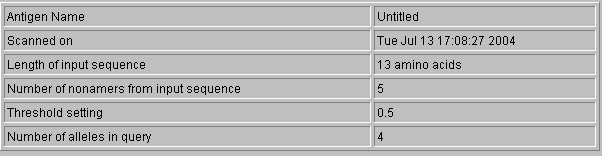
HTML I:-
This format display all overlapping predicted MHC binders in separate lines.This is very useful in detecting the overlapping MHC binders.The display provides a clear indication about the exact position of predicted binder.An example is shown below.
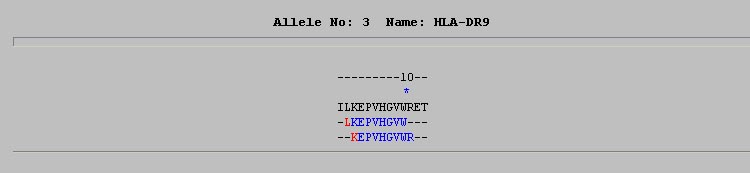
HTML II:-
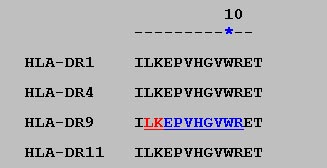
This format dispalys all predicted binder for specific MHC allele in the single line just by coloring the predicted binders.The starting residue of each predicted binder is shown is red and while rest of the residues in blue color.The option is very useful in detecting the promiscious MHC binder in the sequence.The example of HTML II display format is shown below.
Tabular:-
This one is the most common display format used by most prediction methods.The peptides are displayed in table in desending order of their score.The predicted binders of each MHC allele are shown in seprate tablesThe server also provides the facility to customise the number of the top scorer peptides to be displayed in each table.
