MHC2Pred Information
For development of MHC binder prediction method, an elegant machine learning technique SVM (Joachims, 1999; Cristianini and Shawe-Taylor, 2000) has been used.SVM has been trained on the binary input of single amino acid sequence. Each amino acid of 9mer peptide was represented by a 20-dimensional vector. Each peptide of nine amino acids has been represented through a vector of 180 dimensions. The binders have been represented by the +1 and non-binders by -1. A suitable type of kernel for classifying the data has been chosen by conducting experiments with every kernel type i.e. RBF, Polynomial, linear and Sigmoid. The kernel features and regulatory parameter C were optimized by systematic variation in the parameters and evaluations of prediction performance. The overall architecture of SVM based methods is shown in below.
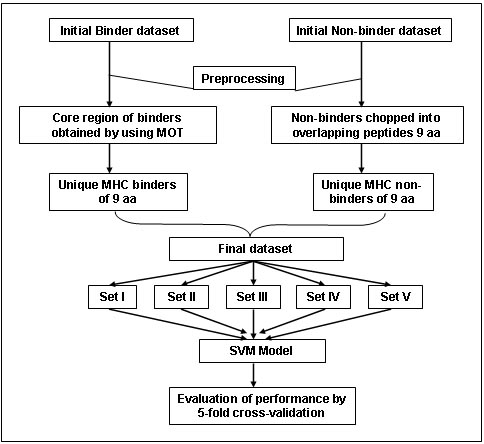
Cross-validation and Performance measures:- The main goal of machine learning approach is to obtain good classification performance on unseen data. Therefore, performance of methods for all alleles has been evaluated using 5-fold cross validation (Kaur and Raghava, 2003). In 5-fold cross-validation the dataset is randomly divided into five equal sized subsets. The method is trained 5 times using 1 distinct set for testing and remaining 4 sets for training. The final performence of the method is obtained by averging. The performance of method has been measured through threshold dependent parameters such as sensitivity, specificity, NPV, PPV and accuracy.