| |
HLP: A webserver for predicting half-life of peptides in intestine like environment |
| Home | Submit: Peptide, Protein, Batch | Data sets | Algorithm | Help | Links | Team | Contact us |
Predict peptide half-life and antibacterial activity in batch mode.
Simultaneously, a number of peptides can be submitted to predict their half-life and antibacterial activity.
Help
Input Sequence:Input Sequence for Peptide/Protein Module(s): Peptide/Protein sequence(s) should be pasted directly (in the relevant text input box provided) in single letter code of amino acids (in single line only). Any unusual character like new line, > sign etc. can lead to an error.Input Sequence for Batch Module: This module accepts the peptide sequences only. The input peptides should be pasted in textbox / uploaded through a text file in FASTA format only. Note: Example input sequences (in form of a button named "Use Example Sequence(s)") are given with each module. Criteria for Model Selection on Peptide, Protein and Batch modulesWe have developed a total of four models to use on our HLP web server. Out of these four, two models have been developed using Support Vector Machine (SVM) and make the use of amino acid composition, dipeptide composition for 16mer, 10mer peptide half-life prediction, respectively. Another two models have been developed using WEKA and make the use of dipeptide composition for selected dipeptides to predict the half life of 10mer and 16mer peptides. It is the user's choice for what length of peptide he/she want to predict half-life, but the selection of model should be in accordance with peptide length (i.e., if selected model is 10mer, the peptide should be minimum 10 residues long or if the selected model is 16mer, submitted peptide should be minimum 16 residues long). The "User-selected sequence length" model is just an additional feature of HLP (not actually a model) that can be used for designing of peptides 5-30 residues long. If user submits a protein/peptide more than 30 residue long, first 30 residues will be used by the Web server/software. Before using "User-selected sequence length" model, please read the flow chart given below: Figure 1: Flow chart showing criteria for model selection.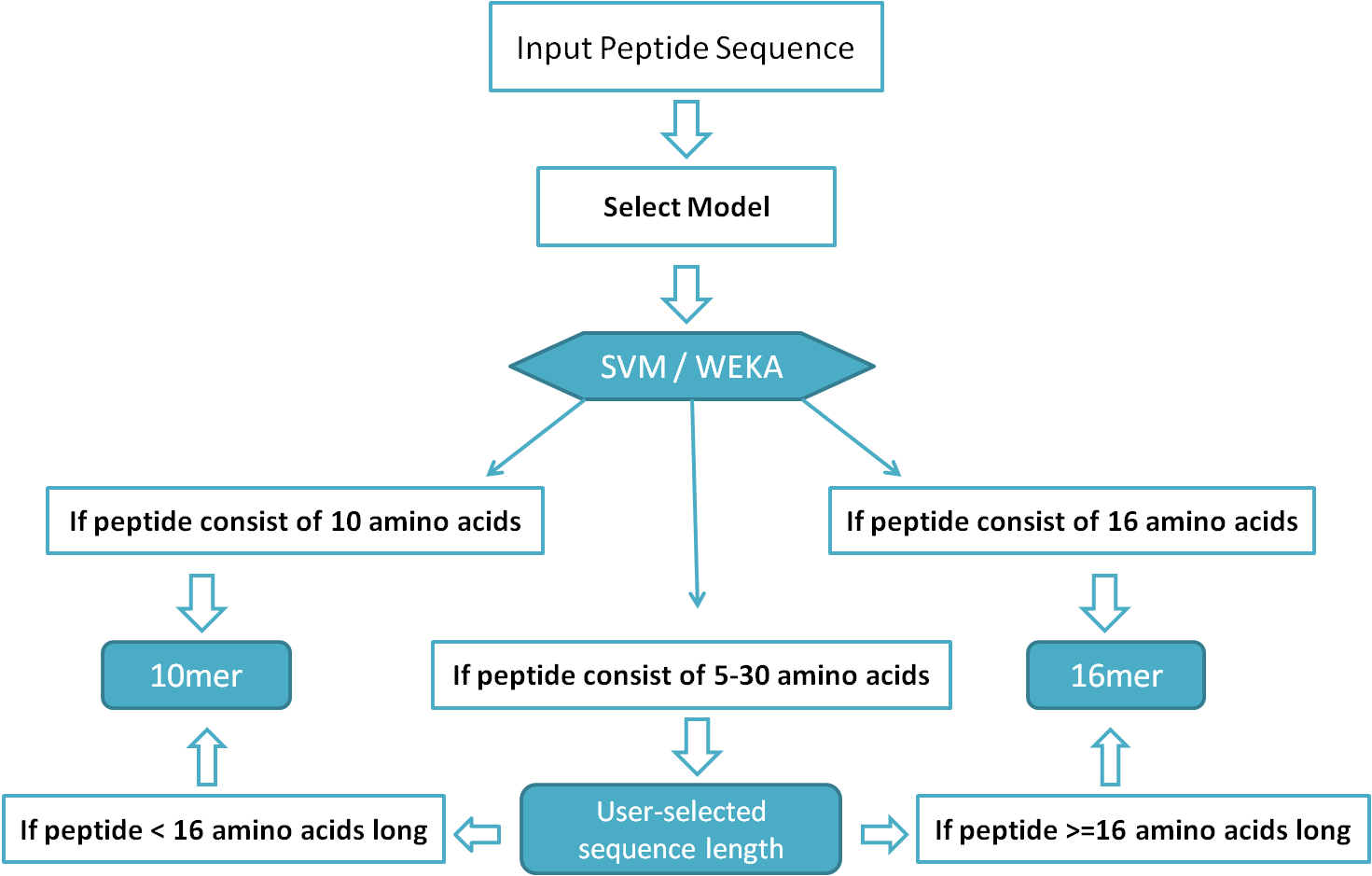
Selection of Model (for Half-life Prediction) for User Selected Peptide Length: Note: This is extrapolation of our models; we cannot validate our models of on variables length of peptides as we have only 10mers and 16mers. (a) If submitted peptide is less than 16 residue long, then half-life is predicted by 10mer model. (b) If submitted peptide is 16 residue or longer, half-life is predicted by 16mer model. Peptide Module:This module permits users to paste their peptide sequences (10mer / 16mer / 5 to 30 residue long) in single letter code of amino acids to design mutants and predict half-life for original and mutant peptides. Additionally, some important physicochemical properties such as hydrophobicity, pKa, pKb, Residue volume, etc. too calculated for original as well as mutant peptides. To optimize the half-life and physicochemical properties, all the mutants may be further mutated through single mouse click over them. The entire procedure can be described through the diagram given below:
Protein Module:This module may be used to scan a protein sequence (of any length, needs to be single lined) to retrieve overlapping peptides (with their predicted half-life and calculated physicochemical properties) from it. Therefore, the module helps in the identification of peptides (with desired half-life and physicochemical properties) from a protein. The entire procedure is described in the diagram given below:
Scanning of Protein Throughout its Length (To generate overlapping peptides)The user submitted protein is scanned throughout its length (in the form of overlapping window) and all possible peptide fragments are generated. Half-life (sec) and physicochemical properties are calculated for these peptides. Thus whole protein is scanned throughout its length and stable peptides are obtained. The figure to show scheme of peptide generation is given below: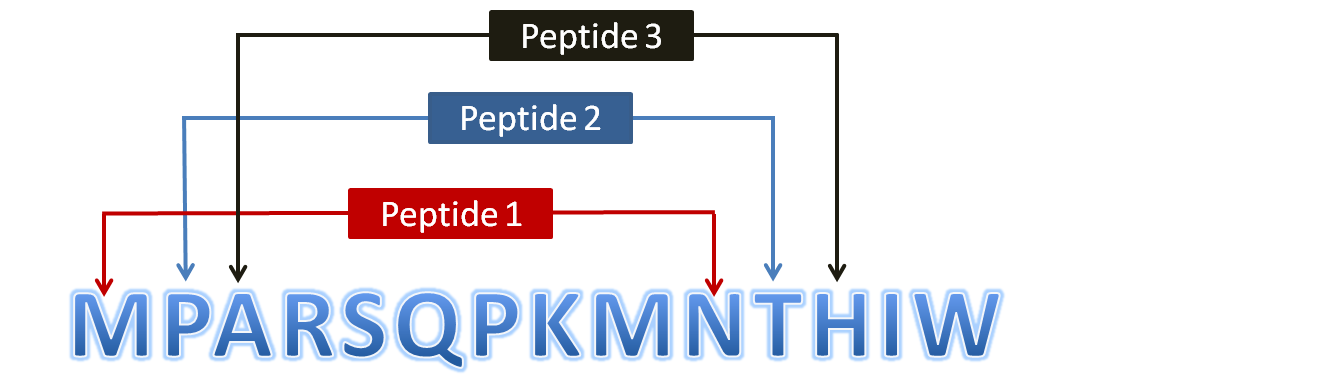
Figure 2: Diagram showing generation of overlapping peptides (10mer) from a user submitted protein sequenceBatch Module:Batch module has been specifically designed for high-throughput screening of peptide sequences and thus helps in prediction of half-life for multiple peptide sequences in one turn rather than submitting again and again through Peptide module. Output results may be displayed in two modes viz; Interactive / Non-interactive mode. Interactive mode allows users to sort their results values in ascending as well as descending orders. Mutation of user submitted peptides is possile just through single mouse click. In contrast to this, non-interactive mode permits to download the results in text format (tab-delimited). These results can be further used to filter various selected values. The usage of Batch module is represented in the diagram given below:

Result TableResults of your query are shown on new page. At this, second column named Peptides is clickable and single click action will generate mutants of that particular peptide with desired properties. First row (with S.No. "0") is always the user input prediction.Description of fields in result table
|