AVP-PEpred: Antiviral Peptide-Percent Efficacy Predictor

Algorithm
Machine learning Techniques:
Support vector machines (SVMs) were trained with the selected sequence features to predict peptide potency in regression mode. SVM allows choosing a number of parameters and kernels. The SVMlight software package (available at http://svmlight.joachims.org/) was used to construct SVM classifiers. In this study, we used the radial basis function (RBF) kernel:
k(x ,y)=exp(-γ||x - y||2)
where x and y are two data vectors, and γ is a training parameter.In addition we also used another machine learning method, Random Forest (R package), but SVM slighly out-performed RF. Random forests are an ensemble learning method for classification (and regression) that operate by constructing a multitude of decision trees at training time and outputting the class that is the mode of the classes output by individual trees. Features:
a) Composition
Amino acid composition is the fraction of each amino acid in a peptide. The fraction of all 20 natural amino acids was calculated using the following equation:
Fraction of amino acid X=Total no. of X/peptide length
b) Database scanning
The sequence segments with high identity are inclined to share the structure and function. This method has been widely used in the past for proteins and peptide prediction, like in signal peptide and antimicrobial peptide prediction using BLASTP. We have also implemented BLASTP algorithm for prediction of AVPs. Each query sequence is matched against two newly released antiviral peptide databases viz. AVPdb and HIPdb described earlier.
c) Physicochemical properties
We tried all 544 physicochemical properties available in AAindex database individually, to look into the importance of each property in antiviral activity of the peptides. Finally selected 15 best performing physicochemical properties were used in developing AVPphysico model as provided below:
QIAN880113 , SUYM030101 , FINA910102 , CHOP780211 , MUNV940105 , TANS770102 , MUNV940104 , PALJ810114 , PALJ810115 , QIAN880110 , AURR980103 , ISOY800104 , MUNV940102 , RACS820110 , and AURR980105.
d) Binary profiles
Binary profiles were generated for the peptides with each amino acid being represented by a vector of 20 dimensions (e.g. Ala by 1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0 and Cys by 0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0 etc .) for the incorporation of positional inforfamation of amino acids in a peptide. A pattern of window length 'w' was represented by a vector of dimensions 20*w.
Evaluation:
In order to evaluate performance of our models, we used Pearson’s correlation coefficient (R). All models were evaluated using ten-fold cross validation technique.
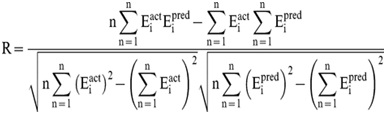
Where n is the size of test set, Eipred and Eiact is the predicted and actual IC50 respectively.
 -Database of antiviral peptides.
-Database of antiviral peptides.